Introduction
The stage of disease and general fitness of patients usually influences the treatments they receive. Because of this, tests of treatments and treatment comparisons need to ensure that treatment comparison groups are made up of patients who are as similar as possible. Without this assurance, any differences in the progress of patients receiving different treatments cannot confidently be ascribed to differential effects of treatments: they may simply reflect differences in the characteristics of patients receiving the different treatments. This is the rationale underlying random allocation (randomisation) to treatment groups in randomised trials – to ensure that whether treatment is given or withheld is unrelated to factors influencing the prognoses of patients.
Sometimes – for one or more of a variety of reasons – the organisation of randomized controlled trials (RCTs) presents challenges that are judged likely to be very difficult to overcome. One such example relates to the need to assess the effects of bone marrow transplantation in the treatment of acute myeloid leukaemia in children. Randomised comparisons of treatment with and without bone marrow transplantation in this form of leukaemia have been judged unlikely to be achieved successfully, yet it is clearly important to obtain reliable, unbiased estimates of the effects of transplantation – wanted and unwanted – because the disadvantages may outweigh any advantages of this invasive treatment.
In 1991, Richard Gray and Keith Wheatley reported an ingenious method for obtaining unbiased estimates of the effects of bone marrow transplantation without conducting a traditional randomised trial (Gray and Wheatley 1991). They pointed out that unbiased comparisons could be made between child patients who had a genetically compatible sibling – and so, in principle, could receive a matched sibling bone marrow transplant – with other child patients who had no genetically compatible sibling – and so were incapable of receiving a matched sibling bone marrow transplant. Because having or not having a genetically compatible sibling is a matter of chance – it is determined by random assortment of genes at the time of gamete formation and conception – this situation produces what is effectively a randomized comparison.
Whether a child with leukaemia belongs to the group with genetically compatible siblings or the group without such siblings will not be related to potential confounding factors such as disease stage and general fitness at the time of diagnosis. In a form of ‘intention-to-treat analysis’, Gray and Wheatley noted that an unbiased comparison meant comparing all the patients with a genetically compatible sibling with all the patients without such a sibling – regardless of whether or not every patient with a potential donor actually received a transplant (see Figure, below).
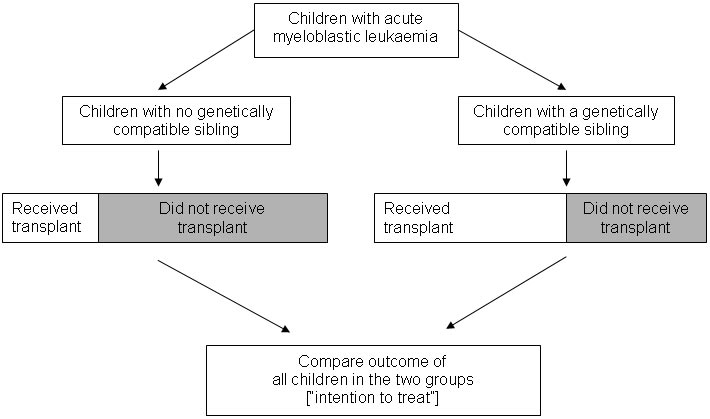
Gray and Wheatley appear to have been the first authors to refer to this particular way of capitalising on chance events in nature to create unbiased comparison groups to assess the effects of a treatment. They referred to it as ‘Mendelian randomisation’. Several further studies have now been carried out using their design (Ljungman et al. 1993; Keating et al. 1998; Burnett et al. 2002), including studies to assess the effects of treatment for acute lymphoblastic leukaemia (Harrison et al. 2000; Bleakley et al. 2002; Balduzzi et al. 2005). Some of these studies have confirmed that like is being compared with like in comparison groups defined in this way (Keating et al. 1998; Harrison et al. 2000; Burnett et al. 2002; Bleakley et al. 2002). They have also shown that there are differences in prognostic factors between groups defined by the treatment they received; differences that would confound a conventional observational analysis comparing different treatments (Harrison et al. 2000; Burnett et al. 2002; Bleakley et al. 2002).
The basic design outlined in the Figure might be improved by taking into account the number of siblings each patient has (Davey Smith and Ebrahim 2003). Patients with more siblings have a greater chance of having a genetically compatible donor, and therefore groups defined by having a compatible donor will differ according to average number of siblings and thus will differ by factors that may be related to prognosis. Indeed, a later study found that the number of siblings could itself be related to survival (Charalampopoulou et al. 2004). The study by Gray and Wheatley (Gray, personal communication) and another more recent study (Burnett et al. 2002) applied an analysis restricted to patients with at least one sibling, but exact stratification or matching on number of siblings might be a more robust approach.
Mendelian randomisation in other settings
Mendelian randomisation as a term has also recently been used to describe methods for obtaining unbiased estimates of causal associations in observational studies in aetiological epidemiology. In part, these developments reflect a response to well-publicised cases of conventional observational epidemiology having produced misleading information about supposed causal or protective factors, for example, for b-carotene and cancer, vitamins C and E and coronary heart disease, and hormone replacement therapy and cardiovascular disease. Observational epidemiological studies suggested that these factors had important protective effects, but randomised controlled trials failed to confirm this. The probable reason for these discrepancies between analyses of observational data and randomised trials is that there is considerable confounding between, for example, dietary vitamin C or E intake or taking b-carotene supplements and various behavioural and socio-economic factors related to increased risks of disease.
Mendelian randomisation – the random assortment of genes from parents to offspring which occurs during gamete formation and conception – provides a method of assessing whether certain environmental exposures are causally related to a disease. The association between risk of a disease and a genetic variant that influences the exposure or mimics the biological link between a proposed exposure and disease is not generally susceptible to the ‘reverse causation’ or confounding that may distort interpretations of conventional observational studies.
The contribution of Martijn Katan
A Dutch researcher, Martijn Katan, was an early exponent of what has since become termed Mendelian randomization. He was concerned that observational studies were suggesting that low serum cholesterol levels were associated with an increased risk of cancer, and thus that treatment to lower cholesterol could have detrimental effects (Garcia-Palmiere et al. 1981). This association might be explained by early cancer lowering cholesterol levels – reverse causality – or by confounding factors (such as cigarette smoking), which are related both to future cancer risk and to lower circulating cholesterol levels (McMichael et al. 1984).
Katan pointed out (Katan 1986) that polymorphic forms of the apolipoprotein ε(APOΕ) gene were related to different average levels of serum cholesterol. If low circulating cholesterol levels were indeed a causal factor for cancer, then individuals with the genotype associated with lower average cholesterol should be expected to have higher cancer risk. If, however, ‘reverse causation’ or confounding generated the association between low cholesterol and cancer, then no association would be expected between APOE genotype and cancer. Individuals with lower cholesterol because of their genotype, rather than because existing clinically unrecognized cancers had lowered their cholesterol, would not have a higher risk of cancer; nor would there be substantial confounding between genotype-associated differences in cholesterol level and lifestyle or socio-economic factors. Although Katan did not have any data to investigate these possibilities, he advocated a study design taking these considerations into account (Katan 1986).
As far as I am aware, Katan’s intriguing suggestion has not yet been explicitly applied to address the important question he posed, although sporadic reports relating APOE to risk of specific cancers continue to appear (Niemi et al. 2000; Moysich et al. 2000; Wessel et al. 2001; Watson et al. 2003; Slattery et al. 2005). However, several examples where the phenotypic effects of polymorphisms are well-documented provide encouraging evidence of the explanatory power of Mendelian randomisation (Davey Smith and Ebrahim 2003; Davey Smith and Ebrahim 2004; Lewis and Davey Smith 2005; Chen et al. 2008).
Why the term “Mendelian randomisation”?
Gregor Mendel (1822-1884) concluded from his hybridisation studies with pea plants that “the behaviour of each pair of differentiating characteristics [such as the shape and colour of seeds] in hybrid union is independent of the other differences between the two original plants” (Mendel 1866). This formulation was actually the only regularity that Mendel referred to as a “law”. In Carl Correns’ 1900 paper (one of a trio appearing that year which are considered to represent the rediscovery of Mendel) he refers to this as Mendel’s Law (Correns 1900; Olby 1966).
Morgan (1913) discusses independent assortment and refers to this process as being realised “whenever two pairs of characters freely Mendelize”. Morgan’s use of Mendel’s surname as a verb did not catch on, but Morgan later christened this as Mendel’s second law (Morgan 1919), and it has been known as this, or as “The Law of Independent assortment”, since this time. The law suggests that inheritance of one trait is independent of – that is, randomized with respect to – the inheritance of other traits.
RA Fisher – simultaneously a founder of statistical genetics and of the application of randomization in experimental studies – clearly linked his two areas of interest in a 1951 lecture (in honour of William Bateson, the rediscoverer of Mendel) on “statistical methods in genetics”. He mentioned “a connection between our two subjects which seem not to be altogether accidental, namely that the “factorial” method of experimentation, now of lively concern so far afield as the psychologists, or the industrial chemists, derives its structure and its name, from the simultaneous inheritance of Mendelian factors. Geneticists certainly need not feel that the intellectual debt is all on one side” (Fisher 1952). He went on to say that Genetics is indeed in a peculiarly favoured condition in that Providence has shielded the geneticist from many of the difficulties of a reliably controlled comparison. The different genotypes possible from the same mating have been beautifully randomised by the meiotic process ……. “Generally speaking the geneticist, even if he foolishly wanted to, could not introduce systematic errors into comparison of genotypes, because for most of the relevant time he has not yet recognised them.” In Fisher’s mind, at least, the factorial randomized trial and genotypic analysis were in someway analogous.
The analogy with a randomized controlled trial will clearly be most applicable to parent-offspring designs investigating the frequency with which one of two alleles from a heterozygous parent is transmitted to offspring with a particular disease. However, at a population level, traits influenced by genetic variants are generally not associated with the social, behavioural and environmental factors that confound relationships observed in conventional epidemiological studies. Thus while the ‘randomization’ is approximate and not absolute in genetic association studies, empirical observations suggest that it applies in most circumstances (Davey Smith et al. 2005; Bhatti et al. 2005; Davey Smith et al. 2008).
As discussed above, the term ‘Mendelian randomisation’ itself was introduced by Gray and Wheatley in a somewhat different context, in which advantage is taken of the random assortment of genetic variants at conception to provide an unconfounded study design for estimating treatment effects for childhood malignancies (Gray and Wheatley 2001; Wheatley and Gray 2004). However, the term has recently become widely used with the meaning ascribed to it here.
The notion that genetic variants can serve as an indicator of the action of environmentally modifiable exposures has been expressed in many contexts. For example, since the mid-1960s various investigators have pointed out that the autosomal dominant condition of lactase persistence is associated with milk drinking. Associations of lactase persistence with osteoporosis, bone mineral density or fracture risk thus provide evidence that milk-drinking protects against these conditions (Birge et al. 1967; Newcomer et al. 1978). In a related vein, it was proposed in 1979 that as N-acetyltransferase pathways are involved in the detoxification of arylamine, a potential bladder carcinogen, the observation of increased bladder cancer risk among people with genetically determined slow acetylator phenotype provided evidence that arylamines are involved in the aetiology of the disease (Lower et al. 1979).
Since that time various commentators have pointed out that the associations of genetic variants of known function with disease outcomes provides evidence about aetiological factors (McGrath et al. 1999; Ames 1999; Rothman et al. 2001; Brennan 2002; Kelada et al. 2003). However, these commentators have not emphasised the key strengths of Mendelian randomization – the avoidance of confounding, bias due to reverse causation or reporting tendency, and the underestimation of risk associations due to variability in behaviours and phenotypes (Davey Smith and Ebrahim 2004).
These key concepts were present in Martijn Katan’s 1986 Lancet letter in which he suggested that genetic variants related to cholesterol level could be used to investigate whether the observed association between low cholesterol and increased cancer risk was real (Katan 1986), and by Honkanen and colleagues in their understanding of how lactase persistence could better characterise the difficult-to-measure environmental influence of calcium intake than could direct dietary reports (Honkanen et al 1996). Since 2000 there have been several reports using the term ‘Mendelian randomization’ in the way it is used here (Youngman et al. 2000; Fallon et al. 2001; Clayton and McKeigue 2001; Keavney 2002; Davey Smith and Ebrahim 2003), and its use is becoming widespread. The fact that ‘Mendelian randomization’ is one of a family of techniques referred to as ‘instrumental variable’ approaches for obtaining robust causal inferences from observational data has also been recognised (Thomas and Conti 2004), and statistical approaches to instrumental variables analysis developed within econometrics have been applied to data from Mendelian randomization studies (Davey Smith et al. 2005; Timpson et al. 2005).
This James Lind Library commentary has been republished in the Journal of the Royal Society of Medicine 2007;100:432-435. Print PDF
Acknowledgements
I am grateful to Richard Gray, Keith Wheatley and Jan Vandenbroucke for comments on earlier drafts of this commentary.
References
Ames BN (1999). Cancer prevention and diet: Help from single nucleotide polymorphisms. Proceedings of the National Academy of Science 96:12216-12218.
Balduzzi A, Valsecchi MG, Uderzo C et al. (2005). Chemotherapy versus allogenic transplantation for very-high-risk childhood acute lymphoblastic leukaemia in first complete remission: comparison by genetic randomisation in an international prospective study. Lancet 366:635-42.
Bhatti P, Sigurdson AL, Wang SS, Chen J, Rothman N, Hartge P, Bergen AW, Landi MT (2005). Genetic variation and willingness to participate in epidemiological research: data from three studies. Cancer Epdemiology Biomarkers and Prevention 14: 2449-53.
Birge SJ, Keutmann HT, Cuatrecasas P, Whedon GD (1967). Osteoporosis, intestinal lactase deficiency and low dietary calcium intake. New England Journal of Medicine 276:445-448.
Bleakley M, Shaw PJ, Nielsen JM (2002). Allogenic bone marrow transplantation for childhood relapsed acute lymphoblastic leukaemia: comparison of outcome in patients with and without a matched family donor. Bone Marrow Transplantation 31:1-7.
Brennan P (2002). Gene environment interaction and aetiology of cancer: what does it mean and how can we measure it? Carcinogenesis 23:381-387.
Burnett AK, Wheatley K, Goldstone AH et al. (2002). The value of allogenic bone marrow transplant in patients with acute myeloid leukaemia at differing risk of relapse: results of the UK MRC AML 10 trial. British Journal of Haematology 118:385-400.
Charalampopoulou A, Petridou E, Spyridopoulos T, Dessypris N, Oikonomou A, Athanasiadou-Piperopoulou F, Baka M, Kalmanti M, Polychronopoulou S, Trichopoulos D (2004). An integrated evaluation of socioeconomic and clinical factors in the survival from childhood acute lymphoblastic leukaemia: a study in Greece. European Journal of Cancer Prevention 13:397-401.
Chen L, Davey Smith G, Harbord R, Lewis S (2008). Alcohol intake and blood pressure: a systematic review implementing Mendelian Randomization approach. PLoS Medicine 5:461-471.
Clayton D, McKeigue PM (2001). Epidemiological methods for studying genes and environmental factors in complex diseases. Lancet 358:1556-60.
Correns C (1900). G. Mendel’s Regel über das Verhalten der Nachkommenschaft der Bastarde. Berichte der Deutschen Botanischen Gesellschaft 8:158-68. [English translation, G. Mendel’s law concerning the behavior of progeny of varietal hybrids, in: Stern and Sherwood;119-32. WH Freeman and Co., USA 1966].
Davey Smith G, Ebrahim S (2003). ‘Mendelian randomization’: can genetic epidemiology contribute to understanding environmental determinants of disease? International Journal of Epidemiology 32:1-22.
Davey Smith G, Ebrahim S (2004). Mendelian randomization: prospects, potentials, and limitations. International Journal of Epidemiology 33:30-42.
Davey Smith G, Ebrahim G (2005). Folate supplementation and cardiovascular disease. Lancet 366:1679-1681.
Davey Smith G, Lawlor DA, Harbord R, Timpson NJ, Day I, Ebrahim S (2008). Clustered environments and randomized genes: a fundamental distinction between conventional and genetic epidemiology. PLoS Medicine 4:1985-1992.
Davey Smith G, Lawlor D, Harbord R, Timpson N, Rumley A, Lowe G, Day I, Ebrahim S (2005). Association of C-reactive protein with blood pressure and hypertension: lifecourse confounding and Mendelian randomisation tests of causality. Arteriosclerosis, Thrombosis, and Vascular Biology 25:1051-1056.
Fallon UB, Ben-Shlomo Y, Elwood P, Ubbink JB, Davey Smith G (2001). Homocysteine and coronary heart disease – Author’s reply. Heart, published online March 14 (http://heart.bmj.com/content/85/2/153.responses#homocysteine-and-coronary-heart-disease—authors-reply).
Fisher RA (1952). Statistical methods in genetics. Heredity 6:1-12.
Garcia-Palmiere MR, Sorlie PD, Costas R, Havlik RJ (1981). An apparent inverse relationship between serum cholesterol and cancer mortality in Puerto Rico. American Journal of Epidemiology 114:29-40.
Gray R, Wheatley K (1991). How to avoid bias when comparing bone marrow transplantation with chemotherapy. Bone Marrow Transplantation 7, Suppl 3:9-12.
Harrison G, Richards S, Lawson S, Darbyshire P, Pinkerton R, Stevens R, Oakhill A, Eden OB. On behalf of the MRC Childhood Leukaemia Working Party (2000). Comparison of allogeneic transplant versus chemotherapy for relapsed childhood acute lymphoblastic leukaemia in the MRC UKALL R1 trial. Annals of Oncology 11:999-1006.
Honkanen R, Pulkkinen P, Järvinen R, Kröger H, Lindstedt K, Tuppurainen M, Uusitupa M (1996). Does lactose intolerance predispose to low bone density? A population-based study of perimenopausal Finnish women. Bone 19:23-28.
Katan MB (1986). Apolipoprotein E isoforms, serum cholesterol, and cancer. Lancet 1:507–08.
Keating S, de Witte T, Suciu S, Willemze R, Hayat M, Labar B, Resegotti L, Ferrini PR, Caronia F, Dardenne M, Solbu G, Petti MC, Vegna ML, Mandelli F, Zittoun RA (1998). The influence of HLA-matched sibling donor availability on treatment outcome for patients with AML: an analysis of the AML 8A study of the EORTC Leukaemia Cooperative Group and GIMENA. British Journal of Haematology 102:1344-53.
Keavney B (2002). Genetic epidemiological studies of coronary heart disease. International Journal of Epidemiology 31:730-36.
Kelada SN, Eaton DL, Wang SS, Rothman NR, Khoury MJ (2003). The role of genetic polymorphisms in environmental health. Environmental Health Perspectives 111:1055-64.
Lawlor DA, Davey Smith G, Bruckdorfer KR, Kundu D, Ebrahim S (2004). Those confounded vitamins: what can we learn from the differences between observational versus randomised trial evidence? Lancet 363:1724-27.
Lewis S, Davey Smith G (2005). Alcohol, ALDH2 and esophageal cancer: a meta-analysis which illustrates the potentials and limitations of a Mendelian randomization approach. Cancer Epidemiology Biomarkers and Prevention 14:1967-1971.
Ljungman P, De Witte T, Verdonck L, Gahrton G, Freycon F, Gravett P, McCann S,
Morgenstern HG, Nikoskelainen J, Powles R et al. (1993). Bone marrow transplantation for acute myeloblastic leukaemia: an EBMT Leukaemia Working Party prospective analysis from HLA-typing. British Journal of Haematology 84:61-66.
Lower GM, Nilsson T, Nelson CE, Wolf H, Gamsky TE, Bryan GT (1979). N-acetylransferase phenotype and risk in urinary bladder cancer: approaches in molecular epidemiology. Environmental Health Perspectives 29:71-9.
McGrath J (1999). Hypothesis: is low prenatal vitamin D a risk-modifying factor for schizophrenia? Schizophrenia Research 40:173-7.
McMichael AJ, Jensen OM, Parkin DM, Zaridze DG (1984). Dietary and endogenous cholesterol and human cancer. Epidemiology Reviews 6:192-216.
Mendel G (1866). Experiments in plant hybridization .
Morgan TH (1913). Heredity and sex. New York: Columbia University Press.
Morgan TH (1919). Physical basis of heredity. Philadelphia: JB Lipincott Company.
Moysich KB, Freudenheim JL, Baker JA, Ambrosone CB, Bowman ED, Schisterman EF, Vena JE, Shields PG (2000). Apolipoprotein E genetic polymorphism, serum lipoproteins, and breast cancer risk. Molecular Carcinogenesis 27:2-9.
Newcomer AD, Hodgson SF, Douglas MD, Thomas PJ (1978). Lactase deficiency: prevalence in osteoporosis. Annals of Internal Medicine 89:218-220.
Niemi M, Kervinen K, Kiviniemi H, Lukkarinen O, Kyllonen AP, Apaja-Sarkkinen M, Savolainen MJ, Kairaluoma MI, Kesaniemi YA (2000). Apolipoprotein E phenotype, cholesterol and breast and prostate cancer. Journal of Epidemiology and Community Health 54:938-9.
Olby RC (1966). Origins of Mendelism. London: Constable.
Rothman N, Wacholder S, Caporaso NE, Garcia-Closas M, Buetow K, Fraumeni JF (2001). The use of common genetic polymorphisms to enhance the epidemiologic study of environmental carcinogens. Biochimica et Biophysica Acta 1471:C1-C10.
Slattery ML, Sweeney C, Murtaugh M, Ni Ma K, Potter JD, R.Levin T, Samowitz W, Wolff R (2005). Associations between apoE genotype and colon and rectal cancer. Carcinogenesis 26:1422-1429.
Thomas DC, Conti DV (2004). Commentary: The concept of ‘Mendelian Randomization’. International Journal of Epidemiology 33:21-25.
Timpson NJ, Lawlor DA, Harbord RM, Gaunt TR, Day IN, Palmer LJ, Hattersley AT, Ebrahim S, Lowe GD, Rumley A, Davey Smith G (2005). C-reactive protein and its role in metabolic syndrome: mendelian randomisation study. Lancet 366:1954-1959.
Watson MA, Gay L, Stebbings WSL, Speakman CTM, Bingham SA, Loktionov A (2003). Apolipoprotein E gene polymorphism and colorectal cancer: gender-specific modulation of risk and prognosis. Clinical Science 104:537-545.
Wessel N, Liestol K, Maehlen J, Brorson SH (2001). The apolipoprotein E epsilon4 allele is no risk factor for prostate cancer in the Norwegian population. British Journal of Cancer 85:1418.
Wheatley K, Gray R (2004). Commentary: Mendelian randomization – an update on its use to evaluate allogeneic stem cell transplantation in leukaemia. International Journal of Epidemiology 33:15-17.
Youngman LD, Keavney BD, Palmer A et al. (2000). Plasma fibrinogen and fibrinogen genotypes in 4685 cases of myocardial infarction and in 6002 controls: test of causality by ‘Mendelian randomization’. Circulation 102 (Suppl II):31-32.
Davey Smith G (2006). Capitalising on Mendelian randomization to assess the effects of treatments.
© George Davey Smith, MRC Centre for Causal Analyses in Translational Epidemiology, University of Bristol, Canynge Hall, Whiteladies Road, Bristol BS8 2PR, UK. Email: george.davey-smith@bristol.ac.uk
Cite as: Davey Smith G (2006). Capitalising on Mendelian randomization to assess the effects of treatments. JLL Bulletin: Commentaries on the history of treatment evaluation (https://www.jameslindlibrary.org/articles/capitalising-on-mendelian-randomization-to-assess-the-effects-of-treatments/)
Introduction
The stage of disease and general fitness of patients usually influences the treatments they receive. Because of this, tests of treatments and treatment comparisons need to ensure that treatment comparison groups are made up of patients who are as similar as possible. Without this assurance, any differences in the progress of patients receiving different treatments cannot confidently be ascribed to differential effects of treatments: they may simply reflect differences in the characteristics of patients receiving the different treatments. This is the rationale underlying random allocation (randomisation) to treatment groups in randomised trials – to ensure that whether treatment is given or withheld is unrelated to factors influencing the prognoses of patients.
Sometimes – for one or more of a variety of reasons – the organisation of randomized controlled trials (RCTs) presents challenges that are judged likely to be very difficult to overcome. One such example relates to the need to assess the effects of bone marrow transplantation in the treatment of acute myeloid leukaemia in children. Randomised comparisons of treatment with and without bone marrow transplantation in this form of leukaemia have been judged unlikely to be achieved successfully, yet it is clearly important to obtain reliable, unbiased estimates of the effects of transplantation – wanted and unwanted – because the disadvantages may outweigh any advantages of this invasive treatment.
In 1991, Richard Gray and Keith Wheatley reported an ingenious method for obtaining unbiased estimates of the effects of bone marrow transplantation without conducting a traditional randomised trial (Gray and Wheatley 1991). They pointed out that unbiased comparisons could be made between child patients who had a genetically compatible sibling – and so, in principle, could receive a matched sibling bone marrow transplant – with other child patients who had no genetically compatible sibling – and so were incapable of receiving a matched sibling bone marrow transplant. Because having or not having a genetically compatible sibling is a matter of chance – it is determined by random assortment of genes at the time of gamete formation and conception – this situation produces what is effectively a randomized comparison.
Whether a child with leukaemia belongs to the group with genetically compatible siblings or the group without such siblings will not be related to potential confounding factors such as disease stage and general fitness at the time of diagnosis. In a form of ‘intention-to-treat analysis’, Gray and Wheatley noted that an unbiased comparison meant comparing all the patients with a genetically compatible sibling with all the patients without such a sibling – regardless of whether or not every patient with a potential donor actually received a transplant (see Figure, below).
Gray and Wheatley appear to have been the first authors to refer to this particular way of capitalising on chance events in nature to create unbiased comparison groups to assess the effects of a treatment. They referred to it as ‘Mendelian randomisation’. Several further studies have now been carried out using their design (Ljungman et al. 1993; Keating et al. 1998; Burnett et al. 2002), including studies to assess the effects of treatment for acute lymphoblastic leukaemia (Harrison et al. 2000; Bleakley et al. 2002; Balduzzi et al. 2005). Some of these studies have confirmed that like is being compared with like in comparison groups defined in this way (Keating et al. 1998; Harrison et al. 2000; Burnett et al. 2002; Bleakley et al. 2002). They have also shown that there are differences in prognostic factors between groups defined by the treatment they received; differences that would confound a conventional observational analysis comparing different treatments (Harrison et al. 2000; Burnett et al. 2002; Bleakley et al. 2002).
The basic design outlined in the Figure might be improved by taking into account the number of siblings each patient has (Davey Smith and Ebrahim 2003). Patients with more siblings have a greater chance of having a genetically compatible donor, and therefore groups defined by having a compatible donor will differ according to average number of siblings and thus will differ by factors that may be related to prognosis. Indeed, a later study found that the number of siblings could itself be related to survival (Charalampopoulou et al. 2004). The study by Gray and Wheatley (Gray, personal communication) and another more recent study (Burnett et al. 2002) applied an analysis restricted to patients with at least one sibling, but exact stratification or matching on number of siblings might be a more robust approach.
Mendelian randomisation in other settings
Mendelian randomisation as a term has also recently been used to describe methods for obtaining unbiased estimates of causal associations in observational studies in aetiological epidemiology. In part, these developments reflect a response to well-publicised cases of conventional observational epidemiology having produced misleading information about supposed causal or protective factors, for example, for b-carotene and cancer, vitamins C and E and coronary heart disease, and hormone replacement therapy and cardiovascular disease. Observational epidemiological studies suggested that these factors had important protective effects, but randomised controlled trials failed to confirm this. The probable reason for these discrepancies between analyses of observational data and randomised trials is that there is considerable confounding between, for example, dietary vitamin C or E intake or taking b-carotene supplements and various behavioural and socio-economic factors related to increased risks of disease.
Mendelian randomisation – the random assortment of genes from parents to offspring which occurs during gamete formation and conception – provides a method of assessing whether certain environmental exposures are causally related to a disease. The association between risk of a disease and a genetic variant that influences the exposure or mimics the biological link between a proposed exposure and disease is not generally susceptible to the ‘reverse causation’ or confounding that may distort interpretations of conventional observational studies.
The contribution of Martijn Katan
A Dutch researcher, Martijn Katan, was an early exponent of what has since become termed Mendelian randomization. He was concerned that observational studies were suggesting that low serum cholesterol levels were associated with an increased risk of cancer, and thus that treatment to lower cholesterol could have detrimental effects (Garcia-Palmiere et al. 1981). This association might be explained by early cancer lowering cholesterol levels – reverse causality – or by confounding factors (such as cigarette smoking), which are related both to future cancer risk and to lower circulating cholesterol levels (McMichael et al. 1984).
Katan pointed out (Katan 1986) that polymorphic forms of the apolipoprotein ε(APOΕ) gene were related to different average levels of serum cholesterol. If low circulating cholesterol levels were indeed a causal factor for cancer, then individuals with the genotype associated with lower average cholesterol should be expected to have higher cancer risk. If, however, ‘reverse causation’ or confounding generated the association between low cholesterol and cancer, then no association would be expected between APOE genotype and cancer. Individuals with lower cholesterol because of their genotype, rather than because existing clinically unrecognized cancers had lowered their cholesterol, would not have a higher risk of cancer; nor would there be substantial confounding between genotype-associated differences in cholesterol level and lifestyle or socio-economic factors. Although Katan did not have any data to investigate these possibilities, he advocated a study design taking these considerations into account (Katan 1986).
As far as I am aware, Katan’s intriguing suggestion has not yet been explicitly applied to address the important question he posed, although sporadic reports relating APOE to risk of specific cancers continue to appear (Niemi et al. 2000; Moysich et al. 2000; Wessel et al. 2001; Watson et al. 2003; Slattery et al. 2005). However, several examples where the phenotypic effects of polymorphisms are well-documented provide encouraging evidence of the explanatory power of Mendelian randomisation (Davey Smith and Ebrahim 2003; Davey Smith and Ebrahim 2004; Lewis and Davey Smith 2005; Chen et al. 2008).
Why the term “Mendelian randomisation”?
Gregor Mendel (1822-1884) concluded from his hybridisation studies with pea plants that “the behaviour of each pair of differentiating characteristics [such as the shape and colour of seeds] in hybrid union is independent of the other differences between the two original plants” (Mendel 1866). This formulation was actually the only regularity that Mendel referred to as a “law”. In Carl Correns’ 1900 paper (one of a trio appearing that year which are considered to represent the rediscovery of Mendel) he refers to this as Mendel’s Law (Correns 1900; Olby 1966).
Morgan (1913) discusses independent assortment and refers to this process as being realised “whenever two pairs of characters freely Mendelize”. Morgan’s use of Mendel’s surname as a verb did not catch on, but Morgan later christened this as Mendel’s second law (Morgan 1919), and it has been known as this, or as “The Law of Independent assortment”, since this time. The law suggests that inheritance of one trait is independent of – that is, randomized with respect to – the inheritance of other traits.
RA Fisher – simultaneously a founder of statistical genetics and of the application of randomization in experimental studies – clearly linked his two areas of interest in a 1951 lecture (in honour of William Bateson, the rediscoverer of Mendel) on “statistical methods in genetics”. He mentioned “a connection between our two subjects which seem not to be altogether accidental, namely that the “factorial” method of experimentation, now of lively concern so far afield as the psychologists, or the industrial chemists, derives its structure and its name, from the simultaneous inheritance of Mendelian factors. Geneticists certainly need not feel that the intellectual debt is all on one side” (Fisher 1952). He went on to say that Genetics is indeed in a peculiarly favoured condition in that Providence has shielded the geneticist from many of the difficulties of a reliably controlled comparison. The different genotypes possible from the same mating have been beautifully randomised by the meiotic process ……. “Generally speaking the geneticist, even if he foolishly wanted to, could not introduce systematic errors into comparison of genotypes, because for most of the relevant time he has not yet recognised them.” In Fisher’s mind, at least, the factorial randomized trial and genotypic analysis were in someway analogous.
The analogy with a randomized controlled trial will clearly be most applicable to parent-offspring designs investigating the frequency with which one of two alleles from a heterozygous parent is transmitted to offspring with a particular disease. However, at a population level, traits influenced by genetic variants are generally not associated with the social, behavioural and environmental factors that confound relationships observed in conventional epidemiological studies. Thus while the ‘randomization’ is approximate and not absolute in genetic association studies, empirical observations suggest that it applies in most circumstances (Davey Smith et al. 2005; Bhatti et al. 2005; Davey Smith et al. 2008).
As discussed above, the term ‘Mendelian randomisation’ itself was introduced by Gray and Wheatley in a somewhat different context, in which advantage is taken of the random assortment of genetic variants at conception to provide an unconfounded study design for estimating treatment effects for childhood malignancies (Gray and Wheatley 2001; Wheatley and Gray 2004). However, the term has recently become widely used with the meaning ascribed to it here.
The notion that genetic variants can serve as an indicator of the action of environmentally modifiable exposures has been expressed in many contexts. For example, since the mid-1960s various investigators have pointed out that the autosomal dominant condition of lactase persistence is associated with milk drinking. Associations of lactase persistence with osteoporosis, bone mineral density or fracture risk thus provide evidence that milk-drinking protects against these conditions (Birge et al. 1967; Newcomer et al. 1978). In a related vein, it was proposed in 1979 that as N-acetyltransferase pathways are involved in the detoxification of arylamine, a potential bladder carcinogen, the observation of increased bladder cancer risk among people with genetically determined slow acetylator phenotype provided evidence that arylamines are involved in the aetiology of the disease (Lower et al. 1979).
Since that time various commentators have pointed out that the associations of genetic variants of known function with disease outcomes provides evidence about aetiological factors (McGrath et al. 1999; Ames 1999; Rothman et al. 2001; Brennan 2002; Kelada et al. 2003). However, these commentators have not emphasised the key strengths of Mendelian randomization – the avoidance of confounding, bias due to reverse causation or reporting tendency, and the underestimation of risk associations due to variability in behaviours and phenotypes (Davey Smith and Ebrahim 2004).
These key concepts were present in Martijn Katan’s 1986 Lancet letter in which he suggested that genetic variants related to cholesterol level could be used to investigate whether the observed association between low cholesterol and increased cancer risk was real (Katan 1986), and by Honkanen and colleagues in their understanding of how lactase persistence could better characterise the difficult-to-measure environmental influence of calcium intake than could direct dietary reports (Honkanen et al 1996). Since 2000 there have been several reports using the term ‘Mendelian randomization’ in the way it is used here (Youngman et al. 2000; Fallon et al. 2001; Clayton and McKeigue 2001; Keavney 2002; Davey Smith and Ebrahim 2003), and its use is becoming widespread. The fact that ‘Mendelian randomization’ is one of a family of techniques referred to as ‘instrumental variable’ approaches for obtaining robust causal inferences from observational data has also been recognised (Thomas and Conti 2004), and statistical approaches to instrumental variables analysis developed within econometrics have been applied to data from Mendelian randomization studies (Davey Smith et al. 2005; Timpson et al. 2005).
This James Lind Library commentary has been republished in the Journal of the Royal Society of Medicine 2007;100:432-435. Print PDF
Acknowledgements
I am grateful to Richard Gray, Keith Wheatley and Jan Vandenbroucke for comments on earlier drafts of this commentary.
References
Ames BN (1999). Cancer prevention and diet: Help from single nucleotide polymorphisms. Proceedings of the National Academy of Science 96:12216-12218.
Balduzzi A, Valsecchi MG, Uderzo C et al. (2005). Chemotherapy versus allogenic transplantation for very-high-risk childhood acute lymphoblastic leukaemia in first complete remission: comparison by genetic randomisation in an international prospective study. Lancet 366:635-42.
Bhatti P, Sigurdson AL, Wang SS, Chen J, Rothman N, Hartge P, Bergen AW, Landi MT (2005). Genetic variation and willingness to participate in epidemiological research: data from three studies. Cancer Epdemiology Biomarkers and Prevention 14: 2449-53.
Birge SJ, Keutmann HT, Cuatrecasas P, Whedon GD (1967). Osteoporosis, intestinal lactase deficiency and low dietary calcium intake. New England Journal of Medicine 276:445-448.
Bleakley M, Shaw PJ, Nielsen JM (2002). Allogenic bone marrow transplantation for childhood relapsed acute lymphoblastic leukaemia: comparison of outcome in patients with and without a matched family donor. Bone Marrow Transplantation 31:1-7.
Brennan P (2002). Gene environment interaction and aetiology of cancer: what does it mean and how can we measure it? Carcinogenesis 23:381-387.
Burnett AK, Wheatley K, Goldstone AH et al. (2002). The value of allogenic bone marrow transplant in patients with acute myeloid leukaemia at differing risk of relapse: results of the UK MRC AML 10 trial. British Journal of Haematology 118:385-400.
Charalampopoulou A, Petridou E, Spyridopoulos T, Dessypris N, Oikonomou A, Athanasiadou-Piperopoulou F, Baka M, Kalmanti M, Polychronopoulou S, Trichopoulos D (2004). An integrated evaluation of socioeconomic and clinical factors in the survival from childhood acute lymphoblastic leukaemia: a study in Greece. European Journal of Cancer Prevention 13:397-401.
Chen L, Davey Smith G, Harbord R, Lewis S (2008). Alcohol intake and blood pressure: a systematic review implementing Mendelian Randomization approach. PLoS Medicine 5:461-471.
Clayton D, McKeigue PM (2001). Epidemiological methods for studying genes and environmental factors in complex diseases. Lancet 358:1556-60.
Correns C (1900). G. Mendel’s Regel über das Verhalten der Nachkommenschaft der Bastarde. Berichte der Deutschen Botanischen Gesellschaft 8:158-68. [English translation, G. Mendel’s law concerning the behavior of progeny of varietal hybrids, in: Stern and Sherwood;119-32. WH Freeman and Co., USA 1966].
Davey Smith G, Ebrahim S (2003). ‘Mendelian randomization’: can genetic epidemiology contribute to understanding environmental determinants of disease? International Journal of Epidemiology 32:1-22.
Davey Smith G, Ebrahim S (2004). Mendelian randomization: prospects, potentials, and limitations. International Journal of Epidemiology 33:30-42.
Davey Smith G, Ebrahim G (2005). Folate supplementation and cardiovascular disease. Lancet 366:1679-1681.
Davey Smith G, Lawlor DA, Harbord R, Timpson NJ, Day I, Ebrahim S (2008). Clustered environments and randomized genes: a fundamental distinction between conventional and genetic epidemiology. PLoS Medicine 4:1985-1992.
Davey Smith G, Lawlor D, Harbord R, Timpson N, Rumley A, Lowe G, Day I, Ebrahim S (2005). Association of C-reactive protein with blood pressure and hypertension: lifecourse confounding and Mendelian randomisation tests of causality. Arteriosclerosis, Thrombosis, and Vascular Biology 25:1051-1056.
Fallon UB, Ben-Shlomo Y, Elwood P, Ubbink JB, Davey Smith G (2001). Homocysteine and coronary heart disease – Author’s reply. Heart, published online March 14 (http://heart.bmj.com/content/85/2/153.responses#homocysteine-and-coronary-heart-disease—authors-reply).
Fisher RA (1952). Statistical methods in genetics. Heredity 6:1-12.
Garcia-Palmiere MR, Sorlie PD, Costas R, Havlik RJ (1981). An apparent inverse relationship between serum cholesterol and cancer mortality in Puerto Rico. American Journal of Epidemiology 114:29-40.
Gray R, Wheatley K (1991). How to avoid bias when comparing bone marrow transplantation with chemotherapy. Bone Marrow Transplantation 7, Suppl 3:9-12.
Harrison G, Richards S, Lawson S, Darbyshire P, Pinkerton R, Stevens R, Oakhill A, Eden OB. On behalf of the MRC Childhood Leukaemia Working Party (2000). Comparison of allogeneic transplant versus chemotherapy for relapsed childhood acute lymphoblastic leukaemia in the MRC UKALL R1 trial. Annals of Oncology 11:999-1006.
Honkanen R, Pulkkinen P, Järvinen R, Kröger H, Lindstedt K, Tuppurainen M, Uusitupa M (1996). Does lactose intolerance predispose to low bone density? A population-based study of perimenopausal Finnish women. Bone 19:23-28.
Katan MB (1986). Apolipoprotein E isoforms, serum cholesterol, and cancer. Lancet 1:507–08.
Keating S, de Witte T, Suciu S, Willemze R, Hayat M, Labar B, Resegotti L, Ferrini PR, Caronia F, Dardenne M, Solbu G, Petti MC, Vegna ML, Mandelli F, Zittoun RA (1998). The influence of HLA-matched sibling donor availability on treatment outcome for patients with AML: an analysis of the AML 8A study of the EORTC Leukaemia Cooperative Group and GIMENA. British Journal of Haematology 102:1344-53.
Keavney B (2002). Genetic epidemiological studies of coronary heart disease. International Journal of Epidemiology 31:730-36.
Kelada SN, Eaton DL, Wang SS, Rothman NR, Khoury MJ (2003). The role of genetic polymorphisms in environmental health. Environmental Health Perspectives 111:1055-64.
Lawlor DA, Davey Smith G, Bruckdorfer KR, Kundu D, Ebrahim S (2004). Those confounded vitamins: what can we learn from the differences between observational versus randomised trial evidence? Lancet 363:1724-27.
Lewis S, Davey Smith G (2005). Alcohol, ALDH2 and esophageal cancer: a meta-analysis which illustrates the potentials and limitations of a Mendelian randomization approach. Cancer Epidemiology Biomarkers and Prevention 14:1967-1971.
Ljungman P, De Witte T, Verdonck L, Gahrton G, Freycon F, Gravett P, McCann S,
Morgenstern HG, Nikoskelainen J, Powles R et al. (1993). Bone marrow transplantation for acute myeloblastic leukaemia: an EBMT Leukaemia Working Party prospective analysis from HLA-typing. British Journal of Haematology 84:61-66.
Lower GM, Nilsson T, Nelson CE, Wolf H, Gamsky TE, Bryan GT (1979). N-acetylransferase phenotype and risk in urinary bladder cancer: approaches in molecular epidemiology. Environmental Health Perspectives 29:71-9.
McGrath J (1999). Hypothesis: is low prenatal vitamin D a risk-modifying factor for schizophrenia? Schizophrenia Research 40:173-7.
McMichael AJ, Jensen OM, Parkin DM, Zaridze DG (1984). Dietary and endogenous cholesterol and human cancer. Epidemiology Reviews 6:192-216.
Mendel G (1866). Experiments in plant hybridization .
Morgan TH (1913). Heredity and sex. New York: Columbia University Press.
Morgan TH (1919). Physical basis of heredity. Philadelphia: JB Lipincott Company.
Moysich KB, Freudenheim JL, Baker JA, Ambrosone CB, Bowman ED, Schisterman EF, Vena JE, Shields PG (2000). Apolipoprotein E genetic polymorphism, serum lipoproteins, and breast cancer risk. Molecular Carcinogenesis 27:2-9.
Newcomer AD, Hodgson SF, Douglas MD, Thomas PJ (1978). Lactase deficiency: prevalence in osteoporosis. Annals of Internal Medicine 89:218-220.
Niemi M, Kervinen K, Kiviniemi H, Lukkarinen O, Kyllonen AP, Apaja-Sarkkinen M, Savolainen MJ, Kairaluoma MI, Kesaniemi YA (2000). Apolipoprotein E phenotype, cholesterol and breast and prostate cancer. Journal of Epidemiology and Community Health 54:938-9.
Olby RC (1966). Origins of Mendelism. London: Constable.
Rothman N, Wacholder S, Caporaso NE, Garcia-Closas M, Buetow K, Fraumeni JF (2001). The use of common genetic polymorphisms to enhance the epidemiologic study of environmental carcinogens. Biochimica et Biophysica Acta 1471:C1-C10.
Slattery ML, Sweeney C, Murtaugh M, Ni Ma K, Potter JD, R.Levin T, Samowitz W, Wolff R (2005). Associations between apoE genotype and colon and rectal cancer. Carcinogenesis 26:1422-1429.
Thomas DC, Conti DV (2004). Commentary: The concept of ‘Mendelian Randomization’. International Journal of Epidemiology 33:21-25.
Timpson NJ, Lawlor DA, Harbord RM, Gaunt TR, Day IN, Palmer LJ, Hattersley AT, Ebrahim S, Lowe GD, Rumley A, Davey Smith G (2005). C-reactive protein and its role in metabolic syndrome: mendelian randomisation study. Lancet 366:1954-1959.
Watson MA, Gay L, Stebbings WSL, Speakman CTM, Bingham SA, Loktionov A (2003). Apolipoprotein E gene polymorphism and colorectal cancer: gender-specific modulation of risk and prognosis. Clinical Science 104:537-545.
Wessel N, Liestol K, Maehlen J, Brorson SH (2001). The apolipoprotein E epsilon4 allele is no risk factor for prostate cancer in the Norwegian population. British Journal of Cancer 85:1418.
Wheatley K, Gray R (2004). Commentary: Mendelian randomization – an update on its use to evaluate allogeneic stem cell transplantation in leukaemia. International Journal of Epidemiology 33:15-17.
Youngman LD, Keavney BD, Palmer A et al. (2000). Plasma fibrinogen and fibrinogen genotypes in 4685 cases of myocardial infarction and in 6002 controls: test of causality by ‘Mendelian randomization’. Circulation 102 (Suppl II):31-32.